3loh: Human insulin receptor ectodomain in complex with two F(ab) fragments per monomer
3.8Å crystal, 1666 protein residues (data and original model: McKern et al., 2006; 3loh model based on a later revision by Smith et al., 2010)
(The videos below recapitulate corrections originally made in collaboration with Mike Lawrence and published in Croll et al., 2016. I am very grateful for Mike's help and support throughout this journey).
At the time this crystal was first solved, high-resolution structures of the first three insulin receptor domains and many homologous antibody F(ab) fragments already existed - but no template structure existed for the three fibronectin type-III (FnIII) domains constituting the "leg" of the receptor. These had to be painstakingly hand-built into the density with the aid of published - but contradictory - secondary structure predictions. In the early 2000s (and still today) hand building into density this fuzzy was a task not for the faint of heart! At 3.8Å, most sidechains appear as little more than nondescript bumps on the backbone (or disappear entirely), and loops are typically reduced to featureless tubes. On top of this, the achingly low data:parameter ratio meant that overfitting and model bias (the unavoidable tendency for the maps to always look a bit like the current model) was a constant challenge. As a result, substantial errors in structures first solved at this resolution are a common phenomenon:
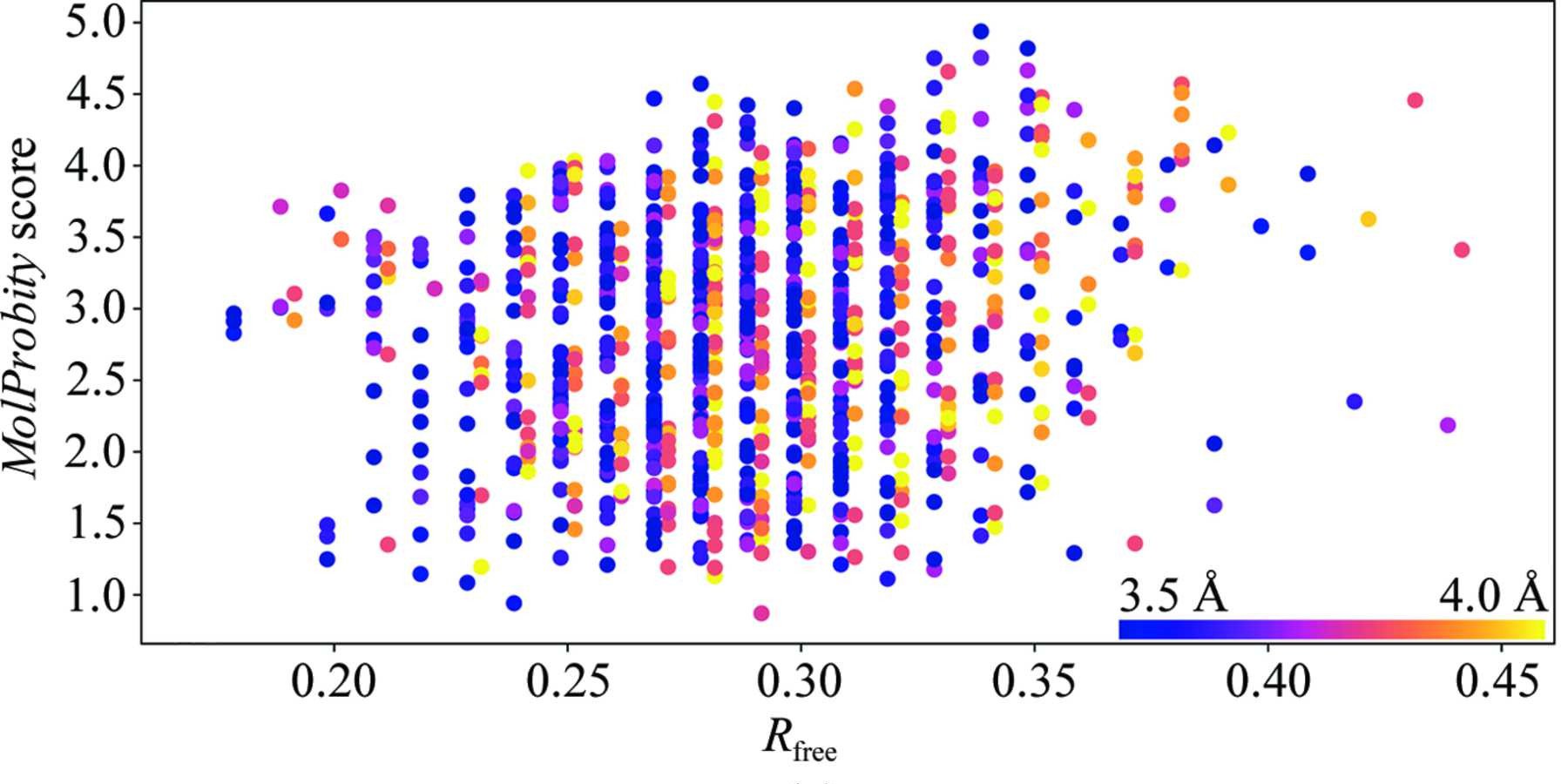
Above: Figure 6a from ISOLDE's inaugural paper (Croll, 2018) plotting rate of stereochemical error as MolProbity score (lower is better) on the y axis against fit to data as Rfree (again, lower is better) for all 3.5-4.0Å crystal structures published between 2006 and October 2016. The first observation to note is that there is little to no correlation between R factors and model quality at these resolutions. The second is that the average error rate is high - keep in mind that the MolProbity score is related to ln(error rate), and a score close to 1 is typical for atomic-resolution models.
Despite the fact that substantial errors arise in many structures of this resolution, this one will always hold a special place in my heart. As discussed in the About section, it was my discovery of the error shown below in the FnIII-3 domain that led me into the endlessly fascinating world of structural biology.
Notes on general model preparation and remodelling
It's a simple trueism that an atomic B-factor (a stand-in for the mobility of an atom, dictating how "smeared out" its density is in the crystal) can only be meaningful if the atom is in the correct place to start with. When rebuilding a low-resolution model, therefore, it is often advantageous to be very conservative with B-factors to reduce overfitting effects. In this case I have removed the original TLS parameterisation for the time being, and reset all atomic B-factors to the Wilson B (about 153Å2 for this crystal). While this initially drives the R-factors on equilibration much higher than when the original B-factor model is used (Rwork/Rfree about 0.36/0.41 vs 0.32/0.36), the potential for model bias (and development of artefacts following large rearrangements) is significantly reduced. Each of the videos below derives from this starting model, and each has the corrections from any previous videos applied. After applying all the corrections shown, restrained refinement in phenix.refine (isotropic B-factors only, no TLS) reduced Rwork/Rfree to 0.258/0.309, with a MolProbity score of 1.86 (down from 4.12). This could undoubtedly be improved further with another round of inspection and rebuilding.
F(ab) chain D - simple 1-residue register shift
This is a good illustration of one of ISOLDE's primary goals: to make simple problems simple to fix. Here we have a short loop (forming one of the complementarity-determining regions) that has been built one residue out of register (meaning that what points inwards should point outwards, and vice versa). Once seen, it's fairly obvious what needs to be done about it, and ISOLDE's register shifter makes short work of the issue.
FnIII-1: large register shifts in two beta strands
Residues 500~540 of the insulin receptor constitute two beta strands and a large intervening loop. The beta strands constitute a substantial portion of a key receptor dimer interface, while the loop as modelled places a number of conserved residues very close to the insulin binding site. Furthermore, new data published in 2018 shows that much of this rregion is directly involved in insulin binding - so correct modelling of it is thus rather important to our understanding of how the receptor behaves. As originally published, the N- and C-terminal strands were out of register by 4 and 6 residues respectively. Correcting them pulls the intervening loop (containing one of the two intermolecular disulfides stabilising the receptor dimer) tighter by ten residues, leaving it buried quite snugly in the dimer interface. The extra residues in turn increase the size of the loops N- and C-terminal to this stretch.
The large register shifts in this domain also strengthen the argument for a very conservative B-factor parameterisation at this stage. Take residues Trp529-Val532 as an example: in the original model these appear as part of a highly flexible loop, with B-factors averaging 314Å2. After remodelling, they are shifted in register by 6 residues to instead form the N-terminus of a well-resolved beta-strand, with B-factors after refining averaging 148Å2. Meanwhile, the residues that were modelled as beta strand are pushed out to become part of a flexible loop. Attempting this shift with the original B-factors creates a result that looks wrong, with strong positive difference density along the beta strand and negative difference density in the following loop. Until such time as ISOLDE gains a robust B-factor refinement algorithm, my advice is to stick to the simplest practicable model until you are confident your atomic coordinates are substantially correct.
FnIII-3: complex register shift and rebuilding
This is the site that started my journey. The combination of difficult density (complicated by non-specific crystal contacts to two symmetry-related F(ab) fragments and two N-glycan sites), a lack of templates and a two-residue sequence error dating back to the original sequencing of the gene used for the study caused substantial headaches for the original crystallographers. On facing this for the very first time in 2013, one of my first challenges was simply finding suitable visualisation modes to make it easier to diagnose wide-ranging problems like this. Rebuilding this region makes use of essentially all the tools available in ISOLDE 1.0b2, and can be achieved in about 20 minutes on a GPU workstation.
The 2018 cryo-EM structure of the insulin-bound receptor reveals that this region also appears likely to be important to insulin binding, albeit in a somewhat different manner to the FnIII-I domain. In the 3loh structure (with no insulin bound) the two FnIII-3 domains of the dimer are far apart in space (forming the points of an inverted "V" shape). Insulin binding triggers a dimerisation of these domains, with the dimerisation surface consisting primarily of the region remodelled in this video.